Proton nuclear magnetic resonance (proton NMR, hydrogen-1 NMR, or 1H NMR) is the application of nuclear magnetic resonance in NMR spectroscopy with respect to hydrogen-1 nuclei within the molecules of a substance, in order to determine the structure of its molecules.[1] In samples where natural hydrogen (H) is used, practically all the hydrogen consists of the isotope 1H (hydrogen-1; i.e. having a proton for a nucleus).

Simple NMR spectra are recorded in solution, and solvent protons must not be allowed to interfere. Deuterated (deuterium = 2H, often symbolized as D) solvents especially for use in NMR are preferred, e.g. deuterated water, D2O, deuterated acetone, (CD3)2CO, deuterated methanol, CD3OD, deuterated dimethyl sulfoxide, (CD3)2SO, and deuterated chloroform, CDCl3. However, a solvent without hydrogen, such as carbon tetrachloride, CCl4 or carbon disulfide, CS2, may also be used.
Historically, deuterated solvents were supplied with a small amount (typically 0.1%) of tetramethylsilane (TMS) as an internal standard for referencing the chemical shifts of each analyte proton. TMS is a tetrahedral molecule, with all protons being chemically equivalent, giving one single signal, used to define a chemical shift = 0 ppm. [2] It is volatile, making sample recovery easy as well. Modern spectrometers are able to reference spectra based on the residual proton in the solvent (e.g. the CHCl3, 0.01% in 99.99% CDCl3). Deuterated solvents are now commonly supplied without TMS.
Deuterated solvents permit the use of deuterium frequency-field lock (also known as deuterium lock or field lock) to offset the effect of the natural drift of the NMR's magnetic field . In order to provide deuterium lock, the NMR constantly monitors the deuterium signal resonance frequency from the solvent and makes changes to the to keep the resonance frequency constant.[3] Additionally, the deuterium signal may be used to accurately define 0 ppm as the resonant frequency of the lock solvent and the difference between the lock solvent and 0 ppm (TMS) are well known.

Proton NMR spectra of most organic compounds are characterized by chemical shifts in the range +14 to -4 ppm and by spin–spin coupling between protons. The integration curve for each proton reflects the abundance of the individual protons.
Simple molecules have simple spectra. The spectrum of ethyl chloride consists of a triplet at 1.5 ppm and a quartet at 3.5 ppm in a 3:2 ratio. The spectrum of benzene consists of a single peak at 7.2 ppm due to the diamagnetic ring current.
Together with carbon-13 NMR, proton NMR is a powerful tool for molecular structure characterization.
Chemical shifts
editChemical shift values, symbolized by δ, are not precise, but typical – they are to be therefore regarded mainly as a reference. Deviations are in ±0.2 ppm range, sometimes more. The exact value of chemical shift depends on molecular structure and the solvent, temperature, magnetic field in which the spectrum is being recorded and other neighboring functional groups. Hydrogen nuclei are sensitive to the hybridization of the atom to which the hydrogen atom is attached and to electronic effects. Nuclei tend to be deshielded by groups that withdraw electron density. Deshielded nuclei resonate at higher δ values, whereas shielded nuclei resonate at lower δ values.
Examples of electron withdrawing substituents are −OH, −OCOR, −OR, −NO2 and halogens. These cause a downfield shift of approximately 2–4 ppm for H atoms on Cα (an aliphatic C atom directly bonded to the substituent in question) and of less than 1–2 ppm for H atoms on Cβ (an aliphatic C atom bonded to Cα). Carbonyl groups, olefinic fragments and aromatic rings contribute sp2 hybridized carbon atoms to an aliphatic chain. This causes a downfield shift of 1–2 ppm at Cα.
Note that labile protons (−OH, −NH2, −SH) have no characteristic chemical shift. However, such resonances can be identified by the disappearance of a peak when reacted with D2O, as deuterium will replace a protium atom. This method is called a D2O shake. Acidic protons may also be suppressed when a solvent containing acidic deuterium ions (e.g. methanol-d4) is used. An alternate method for identifying protons that are not attached to carbons is the heteronuclear single quantum coherence (HSQC) experiment, which correlates protons and carbons that are one bond away from each other. A hydrogen that is not attached to a carbon can be identified because it does not have a crosspeak in the HSQC spectrum.
| Functional group | CH3 | CH2 | CH |
|---|---|---|---|
| CH2R | 0.8 | 1.3 | 1.6 |
| C=C | 1.6 | 2.0 | 2.6 |
| C≡C | 1.7 | 2.2 | 2.8 |
| C6H5 | 2.3 | 2.6 | 2.9 |
| F | 4.3 | 4.4 | 4.8 |
| Cl | 3.0 | 3.4 | 4.0 |
| Br | 2.7 | 3.4 | 4.1 |
| I | 2.2 | 3.2 | 4.2 |
| OH | 3.3 | 3.5 | 3.8 |
| OR | 3.3 | 3.4 | 3.7 |
| OC6H5 | 3.8 | 4.0 | 4.3 |
| OCOR | 3.6 | 4.1 | 5.0 |
| OCOC6H5 | 3.9 | 4.2 | 5.1 |
| OCOCF3 | 4.0 | 4.4 | — |
| CHO | 2.2 | 2.4 | 2.5 |
| COR | 2.1 | 2.2 | 2.6 |
| COOH | 2.1 | 2.3 | 2.6 |
| COOR | 2.0 | 2.3 | 2.5 |
| CONR2 | 2.0 | 2.1 | 2.4 |
| CN | 2.1 | 2.5 | 3.0 |
| NH2 | 2.5 | 2.7 | 3.0 |
| NR2 | 2.2 | 2.4 | 2.8 |
| NRC6H5 | 2.6 | 3.0 | 3.6 |
| NR3+ | 3.0 | 3.1 | 3.6 |
| NHCOR | 2.9 | 3.3 | 3.7 |
| NO2 | 4.1 | 4.2 | 4.4 |
| SR | 2.1 | 2.5 | 3.1 |
| SOR | 2.6 | 3.1 | — |
| =O (aliphatic aldehyde) | — | — | 9.5 |
| =O (aromatic aldehyde) | — | — | 10 |
| M−H (metal hydride) | — | — | −5 to −15 |
Signal intensity
edit
The integrated intensities of NMR signals are, ideally, proportional to the ratio of the nuclei within the molecule.[4] Together with chemical shift and coupling constants, the integrated intensities allow structural assignments. For mixtures, the signal intensities can be used to determine molar ratios. These considerations are valid only when sufficient time is allowed for full relaxation of the affected signals, as determined by their T1 values. A further complication arises from the difficulty of integrating signals of very different line shapes.
Spin–spin couplings
edit
In addition to chemical shift, NMR spectra allow structural assignments by virtue of spin–spin coupling (and integrated intensities). Because nuclei themselves possess a small magnetic field, they influence each other, changing the energy and hence frequency of nearby nuclei as they resonate—this is known as spin–spin coupling. The most important type in basic NMR is scalar coupling. This interaction between two nuclei occurs through chemical bonds, and can typically be seen up to three bonds away (3-J coupling), although it can occasionally be visible over four to five bonds, though these tend to be considerably weaker.
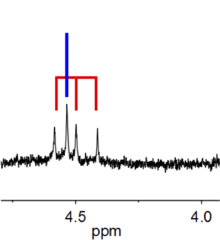
The effect of scalar coupling can be understood by examination of a proton which has a signal at 1 ppm. This proton is in a hypothetical molecule where three bonds away exists another proton (in a CH−CH group, for instance), the neighbouring group (a magnetic field) causes the signal at 1 ppm to split into two, with one peak being a few hertz higher than 1 ppm and the other peak being the same amount lower than 1 ppm. These peaks each have half the area of the former singlet peak. The magnitude of this splitting (difference in frequency between peaks) is known as the coupling constant. A typical coupling constant value for aliphatic protons would be 7 Hz.
The coupling constant is independent of magnetic field strength because it is caused by the magnetic field of another nucleus, not the spectrometer magnet. Therefore, it is quoted in hertz (frequency) and not ppm (chemical shift).
In another molecule a proton resonates at 2.5 ppm and that proton would also be split into two by the proton at 1 ppm. Because the magnitude of interaction is the same, the splitting would have the same coupling constant 7 Hz apart. The spectrum would have two signals, each being a doublet. Each doublet will have the same area because both doublets are produced by one proton each.
The two doublets at 1 ppm and 2.5 ppm from the fictional molecule CH−CH are now changed into CH2−CH:
- The total area of the 1 ppm CH2 peak will be twice that of the 2.5 ppm CH peak.
- The CH2 peak will be split into a doublet by the CH peak—with one peak at 1 ppm + 3.5 Hz and one at 1 ppm − 3.5 Hz (total splitting or coupling constant is 7 Hz).
In consequence the CH peak at 2.5 ppm will be split twice by each proton from the CH2. The first proton will split the peak into two equal intensities and will go from one peak at 2.5 ppm to two peaks, one at 2.5 ppm + 3.5 Hz and the other at 2.5 ppm − 3.5 Hz—each having equal intensities. However, these will be split again by the second proton. The frequencies will change accordingly:
- The 2.5 ppm + 3.5 Hz signal will be split into 2.5 ppm + 7 Hz and 2.5 ppm.
- The 2.5 ppm − 3.5 Hz signal will be split into 2.5 ppm and 2.5 ppm − 7 Hz.
The net result is not a signal consisting of 4 peaks but three: one signal at 7 Hz above 2.5 ppm, two signals occur at 2.5 ppm, and a final one at 7 Hz below 2.5 ppm. The ratio of height between them is 1:2:1. This is known as a triplet and is an indicator that the proton is three-bonds from a CH2 group.
This can be extended to any CHn group. When the CH2−CH group is changed to CH3−CH2, keeping the chemical shift and coupling constants identical, the following changes are observed:
- The relative areas between the CH3 and CH2 subunits will be 3:2.
- The CH3 is coupled to two protons into a 1:2:1 triplet around 1 ppm.
- The CH2 is coupled to three protons.
Something split by three identical protons takes a shape known as a quartet, each peak having relative intensities of 1:3:3:1.
A peak is split by n identical protons into components whose sizes are in the ratio of the nth row of Pascal's triangle:
| n | Name | Row |
|---|---|---|
| 0 | singlet | 1 |
| 1 | doublet | 1 1 |
| 2 | triplet | 1 2 1 |
| 3 | quartet | 1 3 3 1 |
| 4 | quintet | 1 4 6 4 1 |
| 5 | sextet | 1 5 10 10 5 1 |
| 6 | septet | 1 6 15 20 15 6 1 |
| 7 | octet | 1 7 21 35 35 21 7 1 |
| 8 | nonet | 1 8 28 56 70 56 28 8 1 |
Because the nth row has n + 1 components, this type of splitting is said to follow the "n + 1 rule": a proton with n neighbors appears as a cluster of n + 1 peaks.
With 2-methylpropane, (CH3)3CH, as another example: the CH proton is attached to three identical methyl groups containing a total of 9 identical protons. The C−H signal in the spectrum would be split into 10 peaks according to the n + 1 rule of multiplicity. Below are NMR signals corresponding to several simple multiplets of this type. Note that the outer lines of the nonet (which are only 1/8 as high as those of the second peak) can barely be seen, giving a superficial resemblance to a septet.

When a proton is coupled to two different protons, then the coupling constants are likely to be different, and instead of a triplet, a doublet of doublets will be seen. Similarly, if a proton is coupled to two other protons of one type, and a third of another type with a different, smaller coupling constant, then a triplet of doublets is seen. In the example below, the triplet coupling constant is larger than the doublet one. By convention the pattern created by the largest coupling constant is indicated first and the splitting patterns of smaller constants are named in turn. In the case below it would be erroneous to refer to the quartet of triplets as a triplet of quartets. The analysis of such multiplets (which can be much more complicated than the ones shown here) provides important clues to the structure of the molecule being studied.

The simple rules for the spin-spin splitting of NMR signals described above apply only if the chemical shifts of the coupling partners are substantially larger than the coupling constant between them. Otherwise there may be more peaks, and the intensities of the individual peaks will be distorted (second-order effects).
Hetero-nuclear coupling
editIf there are other NMR-active nuclei present in a molecule, spin–spin coupling will be observed between the hetero-atoms and the protons. This occurs most frequently in compounds that contain phosphorus or fluorine, as they are both spin-1/2 nuclei of 100% abundance. For example, the 1H signals for the protons in fluoromethane are split into a doublet by the fluorine atom; conversely, the fluorine-19 NMR spectrum of this compound shows a quartet due to being split by the three protons. Typical 2J coupling constants between fluorine and protons are 48 Hz or so; the strength of coupling declines to 2 Hz in 4J coupling.[5]
Even larger coupling constants may be seen in phosphines, especially if the proton is directly bonded to the phosphorus. Coupling constants for these protons are often as large as 200 Hz, for example, in diethylphosphine, where the 1J P−H coupling constant is 190 Hz.[6] These coupling constants are so large that they may span distances in excess of 1 ppm (depending on the spectrometer), making them prone to overlapping with other proton signals in the molecule.
Carbon satellites and spinning sidebands
editOccasionally, small peaks can be seen shouldering the main 1H NMR peaks. These peaks are not the result of proton-proton coupling, but result from the coupling of 1H atoms to an adjoining carbon-13 (13C) atom. These small peaks are known as carbon satellites as they are small and appear around the main 1H peak i.e. satellite (around) to them. Carbon satellites are small because only very few of the molecules in the sample have that carbon as the rare NMR-active 13C isotope. As always for coupling due to a single spin-1/2 nucleus, the signal splitting for the H attached to the 13C is a doublet. The H attached to the more abundant 12C is not split, so it is a large singlet. The net result is a pair of evenly spaced small signals around the main one. If the H signal would already be split due to H–H coupling or other effects, each of the satellites would also reflect this coupling as well (as usual for complex splitting patterns due to dissimilar coupling partners). Other NMR-active nuclei can also cause these satellites, but carbon is most common culprit in the proton NMR spectra of organic compounds.
Sometimes other peaks can be seen around 1H peaks, known as spinning sidebands and are related to the rate of spin of an NMR tube. These are experimental artifacts from the spectroscopic analysis itself, not an intrinsic feature of the spectrum of the chemical and not even specifically related to the chemical or its structure.
Carbon satellites and spinning sidebands should not be confused with impurity peaks.[7]
See also
edit- Mass spectrometry
- Pople Notation – letter designations for coupled spin-systems
- Nuclear magnetic resonance spectroscopy of proteins
References
edit- ^ R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric Identification of Organic Compounds, 5th Ed., Wiley, 1991.
- ^ "ChemicalShift". Archived from the original on 2016-03-06.
- ^ US patent 4110681, Donald C. Hofer; Vincent N. Kahwaty; Carl R. Kahwaty, "NMR field frequency lock system", issued 1978-08-29
- ^ Balci, M., in "Basic 1H- and 13C-NMR Spectroscopy" (1st Edition, Elsevier), ISBN 978-0444518118.
- ^ "Coupling of Protons with Fluorine Page" (PDF).
- ^ Baccolini, Graziano; Boga, Carla; Mazzacurati, Marzia; Sangirardi, Federico (2006-04-01). "High Atom-Economical One-Pot Synthesis of Secondary Phosphines and Their Borane Complexes Using Recycling Phosphorus Donor Reagent". Organic Letters. 8 (8): 1677–1680. doi:10.1021/ol060284d. ISSN 1523-7060. PMID 16597139.
- ^ Gottlieb HE; Kotlyar V; Nudelman A (October 1997). "NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities". J. Org. Chem. 62 (21): 7512–7515. doi:10.1021/jo971176v. PMID 11671879.
External links
edit- 1H-NMR Interpretation Tutorial
- Spectral Database for Organic Compounds
- Proton Chemical Shifts
- Extensive set of educational examples
- [1] 1D Proton NMR] 1D NMR experiment