Organoiron chemistry is the chemistry of iron compounds containing a carbon-to-iron chemical bond.[1][2] Organoiron compounds are relevant in organic synthesis as reagents such as iron pentacarbonyl, diiron nonacarbonyl and disodium tetracarbonylferrate. Although iron is generally less active in many catalytic applications, it is less expensive and "greener" than other metals.[3] Organoiron compounds feature a wide range of ligands that support the Fe-C bond; as with other organometals, these supporting ligands prominently include phosphines, carbon monoxide, and cyclopentadienyl, but hard ligands such as amines are employed as well.
Iron(–II) and Iron(0)
edit
Carbonyl complexes
editImportant iron carbonyls are the three neutral binary carbonyls, iron pentacarbonyl, diiron nonacarbonyl, and triiron dodecacarbonyl. One or more carbonyl ligands in these compounds can be replaced by a variety of other ligands including alkenes and phosphines. An iron(–II) complex, disodium tetracarbonylferrate (Na2[Fe(CO)4]), also known as "Collman's Reagent," is prepared by reducing iron pentacarbonyl with metallic sodium. The highly nucleophilic anionic reagent can be alkylated and carbonylated to give the acyl derivatives that undergo protonolysis to afford aldehydes:[4]
- LiFe(CO)4(C(O)R) + H+ → RCHO (+ iron containing products)
Similar iron acyls can be accessed by treating iron pentacarbonyl with organolithium compounds:
- ArLi + Fe(CO)5 → LiFe(CO)4C(O)Ar
In this case, the carbanion attacks a CO ligand. In a complementary reaction, Collman's reagent can be used to convert acyl chlorides to aldehydes. Similar reactions can be achieved with [HFe(CO)4]− salts.[5]
Alkene-Fe(0)-CO derivatives
editiron-tricarbonyl-3D-balls.png)
Monoalkenes
editIron pentacarbonyl reacts photochemically with alkenes to give Fe(CO)4(alkene).[6]
Diene-Fe(0)-CO derivatives
editIron diene complexes are usually prepared from Fe(CO)5 or Fe2(CO)9. Derivatives are known for common dienes like cyclohexadiene,[7] norbornadiene and cyclooctadiene, but even cyclobutadiene can be stabilized. In the complex with butadiene, the diene adopts a cis-conformation. Iron carbonyls are potential protective groups for dienes, shielding them from hydrogenations and Diels-Alder reactions. Cyclobutadieneiron tricarbonyl is prepared from 3,4-dichlorocyclobutene and Fe2(CO)9.
Cyclohexadienes, many derived from Birch reduction of aromatic compounds, form derivatives (diene)Fe(CO)3. The affinity of the Fe(CO)3 unit for conjugated dienes is manifested in the ability of iron carbonyls catalyse the isomerisations of 1,5-cyclooctadiene to 1,3-cyclooctadiene. Cyclohexadiene complexes undergo hydride abstraction to give cyclohexadienyl cations, which add nucleophiles. Hydride abstraction from cyclohexadiene iron(0) complexes gives ferrous derivatives.[8][9]
The enone complex (benzylideneacetone)iron tricarbonyl serves as a source of the Fe(CO)3 subunit and is employed to prepare other derivatives. It is used similarly to Fe2(CO)9.
Alkyne-Fe(0)-CO derivatives
editAlkynes react with iron carbonyls to give a large variety of derivatives. Derivatives include ferroles (Fe2(C4R4)(CO)6), (p-quinone)Fe(CO)3, (cyclobutadiene)Fe(CO)3 and many others.[10]
Tri- and polyene Fe(0) complexes
editStable iron-containing complexes with and without CO ligands are known for a wide variety of polyunsaturated hydrocarbons, e.g. cycloheptatriene, azulene, and bullvalene. In the case of cyclooctatetraene (COT), derivatives include Fe(COT)2,[11] Fe3(COT)3,[12] and several mixed COT-carbonyls (e.g. Fe(COT)(CO)3 and Fe2(COT)(CO)6).
2.svg)
Iron(I) and iron(II)
editAs Fe(II) is a common oxidation state for Fe, many organoiron(II) compounds are known. Fe(I) compounds often feature Fe-Fe bonds, but exceptions occur, such as [Fe(anthracene)2]−.[13]
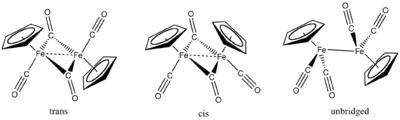
cyclopentadienyliron dicarbonyl dimer

Ferrocene and its derivatives
editThe rapid growth of organometallic chemistry in the 20th century can be traced to the discovery of ferrocene, a very stable compound which foreshadowed the synthesis of many related sandwich compounds. Ferrocene is formed by reaction of sodium cyclopentadienide with iron(II) chloride:
- 2 NaC5H5 + FeCl2 → Fe(C5H5)2 + 2 NaCl
Ferrocene displays diverse reactivity localized on the cyclopentadienyl ligands, including Friedel–Crafts reactions and lithation. Some electrophilic functionalization reactions, however, proceed via initial attack at the Fe center to give the bent [Cp2Fe–Z]+ species (which are formally Fe(IV)). For instance, HF:PF5 and Hg(OTFA)2, give isolable or spectroscopically observable complexes [Cp2Fe–H]+PF6– and Cp2Fe+–Hg–(OTFA)2, respectively.[14][15][16]
Ferrocene is also a structurally unusual scaffold as illustrated by the popularity of ligands such as 1,1'-bis(diphenylphosphino)ferrocene, which are useful in catalysis.[17] Treatment of ferrocene with aluminium trichloride and benzene gives the cation [CpFe(C6H6)]+. Oxidation of ferrocene gives the blue 17e species ferrocenium. Derivatives of fullerene can also act as a highly substituted cyclopentadienyl ligand.
Fp2, Fp−, and Fp+ and derivatives
editFe(CO)5 reacts with cyclopentadiene to give the dinuclear Fe(I) species cyclopentadienyliron dicarbonyl dimer ([FeCp(CO)2]2), often abbreviated as Fp2. Pyrolysis of Fp2 gives the cuboidal cluster [FeCp(CO)]4.
Very hindered substituted cyclopentadienyl ligands can give isolable monomeric Fe(I) species. For example, Cpi-Pr5Fe(CO)2 (Cpi-Pr5 = i-Pr5C5) has been characterized crystallographically.[18]
Reduction of Fp2 with sodium gives "NaFp", containing a potent nucleophile and precursor to many derivatives of the type CpFe(CO)2R.[19] The derivative [FpCH2S(CH3)2]+ has been used in cyclopropanations.[20] The Fp+ fragment is Lewis acidic and readily forms complexes with ethers, amines, pyridine, etc., as well as alkenes and alkynes in the η2 coordination mode. The complex Fp+(η2-vinyl ether)]+ is a masked vinyl cation.[21] Recently, a methane complex, [Fp(CH4)]+[Al(OC(CF3)3)4]–, was prepared and characterized spectroscopically, using a perfluoroalkoxyaluminate as a non-coordinating counterion and 1,1,1,3,3,3-hexafluoropropane as a non-coordinating solvent.[22]
Fp-R compounds are prochiral, and studies have exploited the chiral derivatives CpFe(PPh3)(CO)acyl.[23]
Alkyl, allyl, and aryl compounds
editThe simple peralkyl and peraryl complexes of iron are less numerous than are the Cp and CO derivatives. One example is tetramesityldiiron.

Compounds of the type [(η3-allyl)Fe(CO)4]+X− are allyl cation synthons in allylic substitution.[6] In contrast, compounds of the type [(η5-C5H5)Fe(CO)2(CH2CH=CHR)] possessing η1-allyl groups are analogous to main group allylmetal species (M = B, Si, Sn, etc.) and react with carbon electrophiles to give allylation products with SE2′ selectivity.[24] Similarly, allenyl(cyclopentadienyliron) dicarbonyl complexes exhibit reactivity analogous to main group allenylmetal species and serve as nucleophilic propargyl synthons.[25]
Sulfur and phosphorus derivatives
editComplexes of the type Fe2(SR)2(CO)6 and Fe2(PR2)2(CO)6 form, usually by the reaction of thiols and secondary phosphines with iron carbonyls.[26] The thiolates can also be obtained from the tetrahedrane Fe2S2(CO)6.
Iron(III)
editAlkylation of FeCl3 with methylmagnesium bromide gives [Fe(CH3)4]–, which is thermally labile.[27] Such compounds may be relevant to the mechanism of Fe-catalyzed cross coupling reactions.[28]
Some organoiron(III) compounds are prepared by oxidation of organoiron(II) compounds. A long-known example being ferrocenium [(C5H5)2Fe]+. Organoiron(III) porphyrin complexes, including alkyl and aryl derivatives, are also numerous.[29][30]

Iron(IV)
edit4.svg)
In Fe(norbornyl)4, Fe(IV) is stabilized by an alkyl ligand that resists beta-hydride elimination.[32] Surprisingly, FeCy4, which is susceptible to beta-hydride elimination, has also been isolated and crystallographically characterized and is stable at –20 °C. The unexpected stability was attributed to stabilizing dispersive forces as well as conformational effects that disfavor beta-hydride elimination.[33]
Two-electron oxidation of decamethylferrocene gives the dication [Fe(C5Me5)2]2+, which forms a carbonyl complex, [Fe(C5Me5)2(CO)](SbF6)2.[34] Ferrocene is also known to undergo protonation at the iron center with HF/AlCl3 or HF/PF5 to give the formally Fe(IV) hydride complex, [Cp2FeH]+[PF6]–.[35][36]
Iron(V, VI, VII)
editIn 2020, Jeremy M. Smith and coworkers reported crystallographically characterized Fe(V) and Fe(VI) bisimido complexes based on a bidentate bis(carbene)borate ligand.[37] By virtue of the supporting ligand architecture, these species constitute organometallic Fe(V) and Fe(VI) complexes.
In 2024, Karsten Meyer and coworkers reported a crystallographically characterized Fe(VI) nitrido complex, [(TIMMNMes)FeVI(≡N)(F)](PF6)2·CH2Cl2, which bears a tris(N-heterocyclic carbene) ligand (tris[(3-mesityl-imidazol-2-ylidene)methyl]amine). Related Fe(V) complexes were crystallographically characterized in the same study, while an Fe(VII) species that decomposes above –50 °C was characterized by Mössbauer spectroscopy.[38]
Organoiron compounds in organic synthesis and homogeneous catalysis
editIn industrial catalysis, iron complexes are seldom used in contrast to cobalt and nickel. Because of the low cost and low toxicity of its salts, iron is attractive as a stoichiometric reagent. Some areas of investigation include:
- Hydrogenation and reduction, for example catalyst Knölker complex.
- Cross-coupling reactions. Iron compounds such as Fe(acac)3 catalyze a wide range of cross-coupling reactions with one substrate an aryl or alkyl Grignard and the other substrate an aryl, alkenyl (vinyl), or acyl organohalide. In the related Kumada coupling the catalysts are based on palladium and nickel.
- Complexes derived from Schiff bases are active catalysts for olefin polymerization.[39]
Biochemistry
editIn the area of bioorganometallic chemistry, organoiron species are found at the active sites of the three hydrogenase enzymes as well as carbon monoxide dehydrogenase.
Further reading
edit- E. A. Koerner von Gustorf; F.-W. Grevels; I. Fischler, eds. (1978). The Organic Chemistry of Iron. Academic Press. doi:10.1016/B978-0-12-417101-5.X5001-X. ISBN 978-0-12-417101-5.
References
edit- ^ Synthesis of Organometallic Compounds: A Practical Guide Sanshiro Komiya Ed. S. Komiya, M. Hurano 1997
- ^ Bolm, Carsten (2004). "Iron-Catalyzed Reactions in Organic Synthesis". Chemical Reviews. 104 (12): 6217–6254. doi:10.1021/cr040664h. PMID 15584700.
- ^ Enthaler, S.; Junge, K.; Beller, M. (2008). "Sustainable Metal Catalysis with Iron: From Rust to a Rising Star?". Angew. Chem. Int. Ed. 47 (18): 3317–3321. doi:10.1002/anie.200800012. PMID 18412184.
- ^ Finke, Richard G.; Sorrell, Thomas N. (1979). "Nucleophilic Acylation with Disodium Tetracarbonylferrate: Methyl 7-oxoheptanoate and Methyl 7-oxoöctanoate". Organic Syntheses. 59: 102. doi:10.15227/orgsyn.059.0102.
- ^ Brunet J. J. (1990). "Tetracarbonylhydridoferrates, MHFe(CO)4: Versatile Tools in Organic Synthesis and Catalysis". Chem. Rev. 90 (1041–1059): 1041. doi:10.1021/cr00104a006.
- ^ a b D. Enders; B. Jandeleit; S. von Berg (2002). "(+)-(1R,2S,3R)-Tetracarbonyl[(1-3η)-1-(Phenylsulfonyl)- But-2-en-1-yl]iron(1+) Tetrafluoroborate". Org. Synth. 78: 189. doi:10.15227/orgsyn.078.0189.
- ^ Pearson, Anthony J.; Sun, Huikai (2008). "Cyclohexadieneiron Tricarbonyl". E-EROS Encyclopedia of Reagents for Organic Synthesis. doi:10.1002/047084289X.rn00791. ISBN 978-0471936237.
- ^ Birch, A. J.; Chamberlain, K. B. (1977). "Tricarbonyl[(2,3,4,5-η)-2,4-cyclohexadien-1-one]iron and Tricarbonyl[(1,2,3,4,5-η)-2-methoxy-2,4-cyclohexadien-1-yl]iron(1+) Hexafluorophosphate(1−) From Anisole". Organic Syntheses. 57: 107. doi:10.15227/orgsyn.057.0107.
- ^ Birch, A. J.; Chamberlain, K. B. (1977). "Alkylation of Dimedone With A Tricarbonyl(Diene)iron Complex: Tricarbonyl[2-[(2,3,4,5-η)-4-methoxy-2,4-cyclohexadien-1-yl]-5,5-dimethyl-1,3-cyclohexanedione]iron". Org. Synth. 57: 16. doi:10.15227/orgsyn.057.0016.
- ^ C. Hoogzand; W. Hubel (1968). "Cyclic Polymerization of Acetylenes by Metal Carbonyl Compounds". In Wender, I.; Pino, P. (eds.). Organic Syntheses via Metal Carbonyls Volume 1. Wiley. ISBN 0-471-93367-8.
- ^ D. H. Gerlach, R. A. Schunn, Inorg. Synth. volume 15, 2 (1974) doi:10.1002/9780470132463.ch1
- ^ Lavallo Vincent, Grubbs Robert H (2009). "Carbenes As Catalysts for Transformations of Organometallic Iron Complexes". Science. 326 (5952): 559–562. Bibcode:2009Sci...326..559L. doi:10.1126/science.1178919. PMC 2841742. PMID 19900894.
- ^ Ellis, J. E. (2019). "The Chatt reaction: conventional routes to homoleptic arenemetalates of d-block elements". Dalton Transactions. 48 (26): 9538–9563. doi:10.1039/C8DT05029E. PMID 30724934. S2CID 73436073.
- ^ Astruc, Didier (2017). "Why is Ferrocene so Exceptional?". European Journal of Inorganic Chemistry. 2017 (1): 6–29. doi:10.1002/ejic.201600983. ISSN 1099-0682.
- ^ Malischewski, Moritz; Seppelt, Konrad; Sutter, Jörg; Heinemann, Frank W.; Dittrich, Birger; Meyer, Karsten (2017). "Protonation of Ferrocene: A Low-Temperature X-ray Diffraction Study of [Cp2FeH](PF6) Reveals an Iron-Bound Hydrido Ligand". Angewandte Chemie International Edition. 56 (43): 13372–13376. doi:10.1002/anie.201704854. ISSN 1521-3773. PMID 28834022.
- ^ Cunningham, Allan F. (1997-03-01). "Mechanism of Mercuration of Ferrocene: General Treatment of Electrophilic Substitution of Ferrocene Derivatives". Organometallics. 16 (6): 1114–1122. doi:10.1021/om960815+. ISSN 0276-7333.
- ^ Petr Stepnicka "Ferrocenes: Ligands, Materials and Biomolecules" J. Wiley, Hoboken, 2008. ISBN 0-470-03585-4
- ^ Sitzmann, Helmut; Dezember, Thomas; Kaim, Wolfgang; Baumann, Frank; Stalke, Dietmar; Kärcher, Joerg; Dormann, Elmar; Winter, Hubert; Wachter, Christoph; Kelemen, Marc (1996). "Synthesis and Characterization of the Stable Dicarbonyl(cyclopentadienyl)iron Radical [(C5R5)Fe(CO)2] (R CHMe2)". Angewandte Chemie International Edition in English. 35 (23–24): 2872–2875. doi:10.1002/anie.199628721. ISSN 1521-3773.
- ^ Keith H. Pannell; Hemant K. Sharma (2010). "(Cyclopentadienyl)dicarbonylmethyliron ((η5-C5H5)Fe(CO)2CH3, FpMe), a Seminal Transition-Metal Alkyl Complex: Mobility of the Methyl Group". Organometallics. 29 (21): 4741–4745. doi:10.1021/om1004594.
- ^ Matthew N. Mattson; Edward J. O'Connor; Paul Helquist (1998). "Cyclopropanation using an Iron-Containing Methylene Transfer Reagent: 1,1-Diphenylcyclopropane". Organic Syntheses; Collected Volumes, vol. 9, p. 372.
- ^ Tony C. T. Chang; Myron Rosenblum; Nancy Simms (1988). "Vinylation Of Enolates with a Vinyl Cation Equivalent: trans-3-Methyl-2-vinylcyclohexanone". Org. Synth. 66: 95. doi:10.15227/orgsyn.066.0095.
- ^ Watson, James D.; Field, Leslie D.; Ball, Graham E. (2022-09-28). "[Fp(CH 4 )] + , [η 5 -CpRu(CO) 2 (CH 4 )] + , and [η 5 -CpOs(CO) 2 (CH 4 )] + : A Complete Set of Group 8 Metal–Methane Complexes". Journal of the American Chemical Society. 144 (38): 17622–17629. doi:10.1021/jacs.2c07124. hdl:1959.4/102418. ISSN 0002-7863.
- ^ Karola Rück-Braun "Iron Acyl Complexes" in Transition Metals for Organic Synthesis. Vol. 1. 2nd Ed., M. Beller, C. Bolm, Eds. Wiley-VCH, 2004, Weinheim. ISBN 3-527-30613-7.
- ^ Cutler, A.; Ehnholt, D.; Lennon, P.; Nicholas, K.; Marten, David F.; Madhavarao, M.; Raghu, S.; Rosan, A.; Rosenblum, M. (1975-05-01). "Chemistry of dicarbonyl .eta.5-cyclopentadienyliron complexes. General syntheses of monosubstituted .eta.2-olefin complexes and of 1-substituted .eta.1-allyl complexes. Conformational effects on the course of deprotonation of (.eta.2-olefin) cations". Journal of the American Chemical Society. 97 (11): 3149–3157. doi:10.1021/ja00844a038. ISSN 0002-7863.
- ^ Wang, Yidong; Zhu, Jin; Durham, Austin C.; Lindberg, Haley; Wang, Yi-Ming (2019-12-18). "α-C–H Functionalization of π-Bonds Using Iron Complexes: Catalytic Hydroxyalkylation of Alkynes and Alkenes". Journal of the American Chemical Society. 141 (50): 19594–19599. doi:10.1021/jacs.9b11716. ISSN 0002-7863. PMID 31791121. S2CID 208611984.
- ^ King, R. B., "Organosulfur Derivatives of Metal Carbonyls. I. The Isolation of Two Isomeric Products in the Reaction of Triiron Dodecacarbonyl with Dimethyl Disulfide", J. Am. Chem. Soc., 1962, 84, 2460.
- ^ Sears, Jeffrey D.; Muñoz, Salvador B.; Cuenca, Maria Camila Aguilera; Brennessel, William W.; Neidig, Michael L. (2019). "Synthesis and Characterization of a Sterically Encumbered Homoleptic Tetraalkyliron(III) Ferrate Complex". Polyhedron. 158: 91–96. doi:10.1016/j.poly.2018.10.041. PMC 6481957. PMID 31031511. and references therein.
- ^ Mako, T. L.; Byers, J. A. (2016). "Recent Advances in Iron-Catalysed Cross Coupling Reactions and Their Mechanistic Underpinning". Inorganic Chemistry Frontiers. 3 (6): 766–790. doi:10.1039/C5QI00295H.
- ^ Ogoshi, Hisanobu; Sugimoto, Hiroshi; Yoshida, Zen-Ichi; Kobayashi, Hanako; Sakai, Hiroshi; Maeda, Yutaka (1982). "Syntheses and magnetic properties of aryliron(III) complexes of octaethylporphyrins". Journal of Organometallic Chemistry. 234 (2): 185–195. doi:10.1016/s0022-328x(00)85854-4. ISSN 0022-328X.
- ^ Lexa, Doris; Mispelter, Joel; Saveant, Jean Michel (1981). "Electroreductive alkylation of iron in porphyrin complexes. Electrochemical and spectral characteristics of .sigma.-alkylironporphyrins". Journal of the American Chemical Society. 103 (23): 6806–6812. doi:10.1021/ja00413a004. ISSN 0002-7863.
- ^ Pascal Doppelt (1984). "Molecular stereochemistry of low-spin five-coordinate phenyl(meso-tetraphenylporphyrinato)iron(III)". Inorg. Chem. 23 (24): 4009–4011. doi:10.1021/ic00192a033.
- ^ B. K. Bower and H. G. Tennent (1972). "Transition metal bicyclo[2.2.1]hept-1-yls". J. Am. Chem. Soc. 94 (7): 2512–2514. doi:10.1021/ja00762a056.
- ^ Casitas, Alicia; Rees, Julian A.; Goddard, Richard; Bill, Eckhard; DeBeer, Serena; Fürstner, Alois (2017-08-14). "Two Exceptional Homoleptic Iron(IV) Tetraalkyl Complexes". Angewandte Chemie International Edition. 56 (34): 10108–10113. doi:10.1002/anie.201612299. PMID 28251752.
- ^ Malischewski, Moritz; Seppelt, Konrad; Sutter, Jörg; Munz, Dominik; Meyer, Karsten (2018). "A Ferrocene-Based Dicationic Iron(IV) Carbonyl Complex". Angewandte Chemie International Edition. 57 (44): 14597–14601. doi:10.1002/anie.201809464. ISSN 1521-3773. PMID 30176109. S2CID 52145802.
- ^ Rosenblum, Myron; Santer, J. O. (1959). "PROTONATION OF FERROCENE BY STRONG ACIDS 1". Journal of the American Chemical Society. 81 (20): 5517–5518. doi:10.1021/ja01529a075. ISSN 0002-7863.
- ^ Malischewski, Moritz; Seppelt, Konrad; Sutter, Jörg; Heinemann, Frank W.; Dittrich, Birger; Meyer, Karsten (2017-10-16). "Protonation of Ferrocene: A Low‐Temperature X‐ray Diffraction Study of [Cp 2 FeH](PF 6 ) Reveals an Iron‐Bound Hydrido Ligand". Angewandte Chemie International Edition. 56 (43): 13372–13376. doi:10.1002/anie.201704854. ISSN 1433-7851.
- ^ Martinez, Jorge L.; Lutz, Sean A.; Yang, Hao; Xie, Jiaze; Telser, Joshua; Hoffman, Brian M.; Carta, Veronica; Pink, Maren; Losovyj, Yaroslav; Smith, Jeremy M. (2020-10-16). "Structural and spectroscopic characterization of an Fe(VI) bis(imido) complex". Science. 370 (6514): 356–359. doi:10.1126/science.abd3054. ISSN 0036-8075.
- ^ Keilwerth, Martin; Mao, Weiqing; Malischewski, Moritz; Jannuzzi, Sergio A. V.; Breitwieser, Kevin; Heinemann, Frank W.; Scheurer, Andreas; DeBeer, Serena; Munz, Dominik; Bill, Eckhard; Meyer, Karsten (2024-01-30). "The synthesis and characterization of an iron(VII) nitrido complex". Nature Chemistry: 1–7. doi:10.1038/s41557-023-01418-4. ISSN 1755-4349. PMC 10997499.
- ^ Allan, L. E. N.; Shaver, M. P.; White, A. J. P. and Gibson, V. C., "Correlation of Metal Spin-State in alpha-Diimine Iron Catalysts with Polymerization Mechanism", Inorg. Chem., 2007, 46, 8963-8970.