Triosephosphate isomerase is an enzyme that in humans is encoded by the TPI1 gene.
| TPI1 | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | TPI1, HEL-S-49, TIM, TPI, TPID, triosephosphate isomerase 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 190450; MGI: 98797; HomoloGene: 128432; GeneCards: TPI1; OMA:TPI1 - orthologs | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||




This gene encodes an enzyme, consisting of two identical proteins, which catalyzes the isomerization of glyceraldehyde 3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP) in glycolysis and gluconeogenesis. Mutations in this gene are associated with triosephosphate isomerase deficiency. Pseudogenes have been identified on chromosomes 1, 4, 6 and 7. Alternative splicing results in multiple transcript variants.[5]
Structure
editTriose Phosphate Isomerase is a member of the alpha and beta (α/β) class of proteins; it is a homodimer, and each subunit contains 247 amino acids. Each TPI1 monomer contains the full set of catalytic residues, but the enzyme is only active in the oligomeric form.[6] Therefore, the enzyme must be in a dimer in order to achieve full function of the enzyme, even though it is not believed that the two active sites participate in cooperativity with each other.[7] Each subunit contains 8 exterior alpha helices surrounding 8 interior beta strands, which form a conserved structural domain called a closed alpha/beta barrel (αβ) or more specifically a TIM barrel. Characteristic of most all TIM barrel domains is the presence of the enzyme's active site in the lower loop regions created by the eight loops that connect the C-termini of the beta strands with the N-termini of the alpha helices. TIM barrel proteins also share a structurally conserved phosphate binding motif, with the phosphate group found in the substrate or cofactors.[5]
In each chain, nonpolar amino acids pointing inward from the beta strands contribute to the hydrophobic core of the structure. The alpha helices are amphipathic: their outer (water-contacting) surfaces are polar, while their inner surfaces are largely hydrophobic.
Function
editTPI catalyzes the transfer of a hydrogen atom from carbon 1 to carbon 2, an intramolecular oxidation-reduction reaction. This isomerization of a ketose to an aldose proceeds through an cis-enediol(ate) intermediate. This isomerization proceeds without any cofactors and the enzyme confers a 109 rate enhancement relative to the nonenzymatic reaction involving a chemical base (acetate ion).[8] In addition to its role in glycolysis, TPI is also involved in several additional metabolic biological processes including gluconeogenesis, the pentose phosphate shunt, and fatty acid biosynthesis.
Clinical significance
editTriosephosphate isomerase deficiency is a disorder characterized by a shortage of red blood cells (anemia), movement problems, increased susceptibility to infection, and muscle weakness that can affect breathing and heart function. The anemia in this condition begins in infancy. Since the anemia results from the premature breakdown of red blood cells (hemolysis), it is known as hemolytic anemia. A shortage of red blood cells to carry oxygen throughout the body leads to extreme tiredness (fatigue), pale skin (pallor), and shortness of breath. When the red cells are broken down, iron and a molecule called bilirubin are released; individuals with triosephosphate isomerase deficiency have an excess of these substances circulating in the blood. Excess bilirubin in the blood causes jaundice, which is a yellowing of the skin and the whites of the eyes. Movement problems typically become apparent by age 2 in people with triosephosphate isomerase deficiency. The movement problems are caused by impairment of motor neurons, which are specialized nerve cells in the brain and spinal cord that control muscle movement. This impairment leads to muscle weakness and wasting (atrophy) and causes the movement problems typical of triosephosphate isomerase deficiency, including involuntary muscle tensing (dystonia), tremors, and weak muscle tone (hypotonia). Affected individuals may also develop seizures. Weakness of other muscles, such as the heart (a condition known as cardiomyopathy) and the muscle that separates the abdomen from the chest cavity (the diaphragm) can also occur in triosephosphate isomerase deficiency. Diaphragm weakness can cause breathing problems and ultimately leads to respiratory failure. Individuals with triosephosphate isomerase deficiency are at increased risk of developing infections because they have poorly functioning white blood cells. These immune system cells normally recognize and attack foreign invaders, such as viruses and bacteria, to prevent infection. The most common infections in people with triosephosphate isomerase deficiency are bacterial infections of the respiratory tract. People with triosephosphate isomerase deficiency often do not survive past childhood due to respiratory failure. In a few rare cases, affected individuals without severe nerve damage or muscle weakness have lived into adulthood.[5] The deficiency is most commonly caused by mutations in TPI1, although mutations in other isoforms have been identified. A common marker for TPI deficiency is the increased accumulation of DHAP in erythrocyte extracts; this is because the defective enzyme no longer has the ability to catalyze the isomerization to GAP. The point mutation does not affect the catalysis rate, but rather, affects the assembly of the enzyme into a homodimer.[9][10]
Recent discoveries in Alzheimer's disease research have indicated that amyloid beta peptide-induced nitro-oxidative damage promotes the nitrotyrosination of TPI in human neuroblastoma cells.[11] Nitrosylated TPI was found to be present in brain slides from double transgenic mice over-expressing human amyloid precursor protein as well as in Alzheimer's disease patients. Specifically, the nitrotyrosination occurs on Tyr164 and Tyr208 within the protein, which are near the center of catalysis; this modification correlates with reduced isomerization activity.
Interactive pathway map
editClick on genes, proteins and metabolites below to link to respective articles.[§ 1]
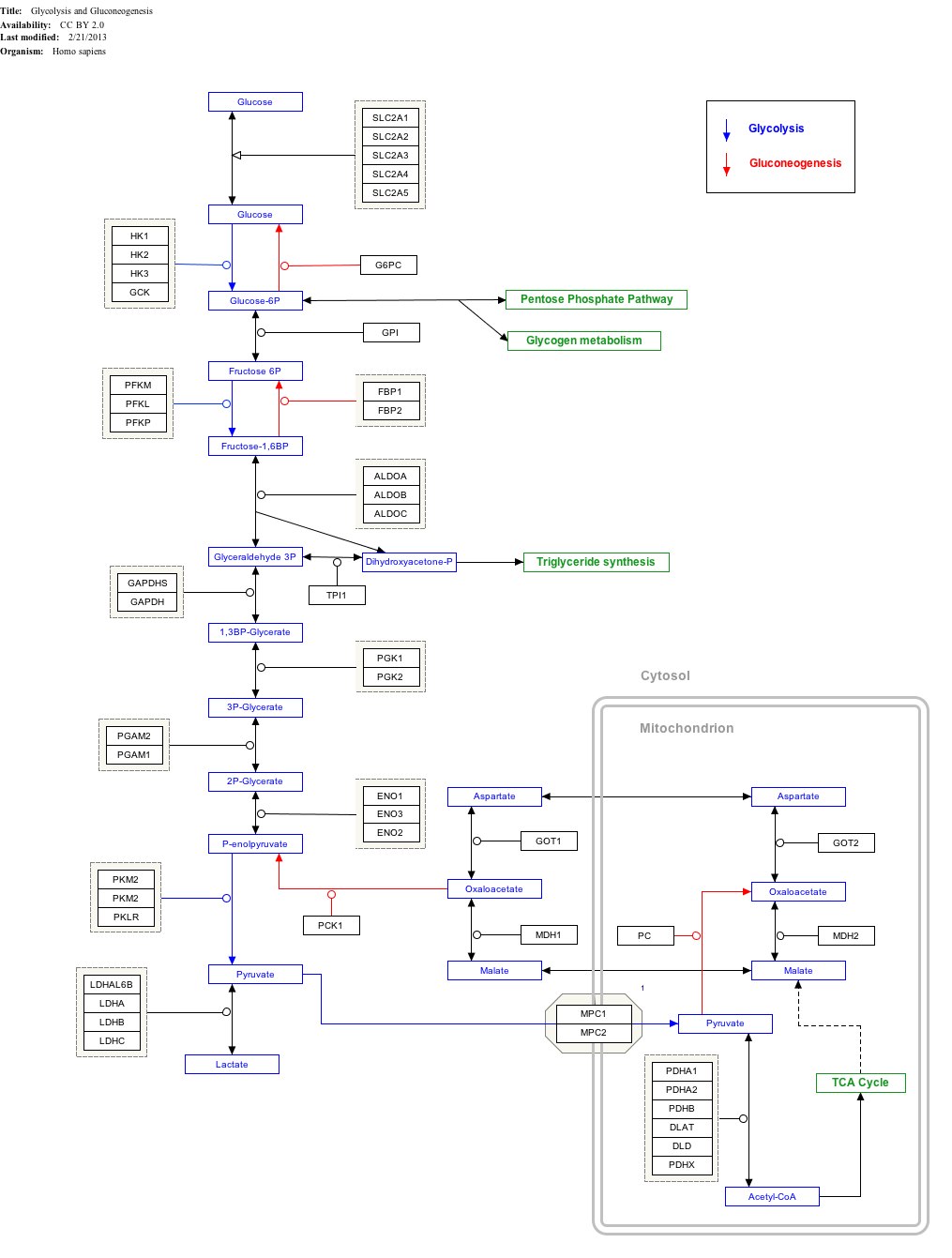
{kind=link}
- ^ The interactive pathway map can be edited at WikiPathways: "GlycolysisGluconeogenesis_WP534".
See also
editReferences
edit- ^ a b c GRCh38: Ensembl release 89: ENSG00000111669 – Ensembl, May 2017
- ^ a b c GRCm38: Ensembl release 89: ENSMUSG00000023456 – Ensembl, May 2017
- ^ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ a b c "Entrez Gene: TPI1 triosephosphate isomerase 1".
- ^ Rodríguez-Almazán C, Arreola R, Rodríguez-Larrea D, Aguirre-López B, de Gómez-Puyou MT, Pérez-Montfort R, Costas M, Gómez-Puyou A, Torres-Larios A (Aug 2008). "Structural basis of human triosephosphate isomerase deficiency: mutation E104D is related to alterations of a conserved water network at the dimer interface". The Journal of Biological Chemistry. 283 (34): 23254–63. doi:10.1074/jbc.M802145200. PMID 18562316.
- ^ Schnackerz KD, Gracy RW (Jul 1991). "Probing the catalytic sites of triosephosphate isomerase by 31P-NMR with reversibly and irreversibly binding substrate analogues". European Journal of Biochemistry. 199 (1): 231–8. doi:10.1111/j.1432-1033.1991.tb16114.x. PMID 2065677.
- ^ Davenport RC, Bash PA, Seaton BA, Karplus M, Petsko GA, Ringe D (Jun 1991). "Structure of the triosephosphate isomerase-phosphoglycolohydroxamate complex: an analogue of the intermediate on the reaction pathway". Biochemistry. 30 (24): 5821–6. doi:10.1021/bi00238a002. PMID 2043623.
- ^ Ralser M, Heeren G, Breitenbach M, Lehrach H, Krobitsch S (20 December 2006). "Triose phosphate isomerase deficiency is caused by altered dimerization--not catalytic inactivity--of the mutant enzymes". PLOS ONE. 1 (1): e30. Bibcode:2006PLoSO...1...30R. doi:10.1371/journal.pone.0000030. PMC 1762313. PMID 17183658.
- ^ Schneider AS (Mar 2000). "Triosephosphate isomerase deficiency: historical perspectives and molecular aspects". Baillière's Best Practice & Research. Clinical Haematology. 13 (1): 119–40. doi:10.1053/beha.2000.0061. PMID 10916682.
- ^ Guix FX, Ill-Raga G, Bravo R, Nakaya T, de Fabritiis G, Coma M, Miscione GP, Villà-Freixa J, Suzuki T, Fernàndez-Busquets X, Valverde MA, de Strooper B, Muñoz FJ (May 2009). "Amyloid-dependent triosephosphate isomerase nitrotyrosination induces glycation and tau fibrillation". Brain. 132 (Pt 5): 1335–45. doi:10.1093/brain/awp023. hdl:10230/60551. PMID 19251756.
Further reading
edit- Ationu A, Humphries A (Dec 1998). "The feasibility of replacement therapy for inherited disorder of glycolysis: triosephosphate isomerase deficiency (review)". International Journal of Molecular Medicine. 2 (6): 701–4. doi:10.3892/ijmm.2.6.701. PMID 9850739.
- Oláh J, Orosz F, Keserü GM, Kovári Z, Kovács J, Hollán S, Ovádi J (Apr 2002). "Triosephosphate isomerase deficiency: a neurodegenerative misfolding disease". Biochemical Society Transactions. 30 (2): 30–8. doi:10.1042/BST0300030. PMID 12023819.
- Rethoré MO, Kaplan JC, Junien C, Lejeune J (Apr 1977). "12pter to 12p12.2: possible assignment of human triose phosphate isomerase". Human Genetics. 36 (2): 235–7. doi:10.1007/BF00273263. PMID 858628. S2CID 25241150.
- Perry BA, Mohrenweiser HW (Mar 1992). "Human triosephosphate isomerase: substitution of Arg for Gly at position 122 in a thermolabile electromorph variant, TPI-Manchester". Human Genetics. 88 (6): 634–8. doi:10.1007/BF02265287. PMID 1339398. S2CID 35721080.
- Dawson SJ, White LA (May 1992). "Treatment of Haemophilus aphrophilus endocarditis with ciprofloxacin". The Journal of Infection. 24 (3): 317–20. doi:10.1016/S0163-4453(05)80037-4. PMID 1602151.
- Boyer TG, Maquat LE (Nov 1990). "Minimal sequence and factor requirements for the initiation of transcription from an atypical, TATATAA box-containing housekeeping promoter". The Journal of Biological Chemistry. 265 (33): 20524–32. doi:10.1016/S0021-9258(17)30534-3. PMID 2243103.
- Maquat LE, Chilcote R, Ryan PM (Mar 1985). "Human triosephosphate isomerase cDNA and protein structure. Studies of triosephosphate isomerase deficiency in man". The Journal of Biological Chemistry. 260 (6): 3748–53. doi:10.1016/S0021-9258(19)83687-6. PMID 2579079.
- Daar IO, Artymiuk PJ, Phillips DC, Maquat LE (Oct 1986). "Human triose-phosphate isomerase deficiency: a single amino acid substitution results in a thermolabile enzyme". Proceedings of the National Academy of Sciences of the United States of America. 83 (20): 7903–7. Bibcode:1986PNAS...83.7903D. doi:10.1073/pnas.83.20.7903. PMC 386831. PMID 2876430.
- Boyer TG, Krug JR, Maquat LE (Mar 1989). "Transcriptional regulatory sequences of the housekeeping gene for human triosephosphate isomerase". The Journal of Biological Chemistry. 264 (9): 5177–87. doi:10.1016/S0021-9258(18)83716-4. PMID 2925688.
- Brown JR, Daar IO, Krug JR, Maquat LE (Jul 1985). "Characterization of the functional gene and several processed pseudogenes in the human triosephosphate isomerase gene family". Molecular and Cellular Biology. 5 (7): 1694–706. doi:10.1128/mcb.5.7.1694. PMC 367288. PMID 4022011.
- Lu HS, Yuan PM, Gracy RW (Oct 1984). "Primary structure of human triosephosphate isomerase". The Journal of Biological Chemistry. 259 (19): 11958–68. doi:10.1016/S0021-9258(20)71304-9. PMID 6434534.
- Mande SC, Mainfroid V, Kalk KH, Goraj K, Martial JA, Hol WG (May 1994). "Crystal structure of recombinant human triosephosphate isomerase at 2.8 A resolution. Triosephosphate isomerase-related human genetic disorders and comparison with the trypanosomal enzyme". Protein Science. 3 (5): 810–21. doi:10.1002/pro.5560030510. PMC 2142725. PMID 8061610.
- Maruyama K, Sugano S (Jan 1994). "Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides". Gene. 138 (1–2): 171–4. doi:10.1016/0378-1119(94)90802-8. PMID 8125298.
- Chang ML, Artymiuk PJ, Wu X, Hollán S, Lammi A, Maquat LE (Jun 1993). "Human triosephosphate isomerase deficiency resulting from mutation of Phe-240". American Journal of Human Genetics. 52 (6): 1260–9. PMC 1682273. PMID 8503454.
- Watanabe M, Zingg BC, Mohrenweiser HW (Feb 1996). "Molecular analysis of a series of alleles in humans with reduced activity at the triosephosphate isomerase locus". American Journal of Human Genetics. 58 (2): 308–16. PMC 1914533. PMID 8571957.
- Mainfroid V, Terpstra P, Beauregard M, Frère JM, Mande SC, Hol WG, Martial JA, Goraj K (Mar 1996). "Three hTIM mutants that provide new insights on why TIM is a dimer". Journal of Molecular Biology. 257 (2): 441–56. doi:10.1006/jmbi.1996.0174. PMID 8609635. S2CID 18248594.
- Ansari-Lari MA, Muzny DM, Lu J, Lu F, Lilley CE, Spanos S, Malley T, Gibbs RA (Apr 1996). "A gene-rich cluster between the CD4 and triosephosphate isomerase genes at human chromosome 12p13". Genome Research. 6 (4): 314–26. doi:10.1101/gr.6.4.314. PMID 8723724.
- Ansari-Lari MA, Shen Y, Muzny DM, Lee W, Gibbs RA (Mar 1997). "Large-scale sequencing in human chromosome 12p13: experimental and computational gene structure determination". Genome Research. 7 (3): 268–80. doi:10.1101/gr.7.3.268. PMID 9074930.
- Rasmussen RK, Ji H, Eddes JS, Moritz RL, Reid GE, Simpson RJ, Dorow DS (1997). "Two-dimensional electrophoretic analysis of human breast carcinoma proteins: mapping of proteins that bind to the SH3 domain of mixed lineage kinase MLK2". Electrophoresis. 18 (3–4): 588–98. doi:10.1002/elps.1150180342. PMID 9150946. S2CID 37336552.
- Ji H, Reid GE, Moritz RL, Eddes JS, Burgess AW, Simpson RJ (1997). "A two-dimensional gel database of human colon carcinoma proteins". Electrophoresis. 18 (3–4): 605–13. doi:10.1002/elps.1150180344. PMID 9150948. S2CID 25454450.