HMG-CoA reductase (3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, official symbol HMGCR) is the rate-controlling enzyme (NADH-dependent, EC 1.1.1.88; NADPH-dependent, EC 1.1.1.34) of the mevalonate pathway, the metabolic pathway that produces cholesterol and other isoprenoids. HMGCR catalyzes the conversion of HMG-CoA to mevalonic acid, a necessary step in the biosynthesis of cholesterol. Normally in mammalian cells this enzyme is competitively suppressed so that its effect is controlled. This enzyme is the target of the widely available cholesterol-lowering drugs known collectively as the statins, which help treat dyslipidemia.







| hydroxymethylglutaryl-CoA reductase (NADH) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC no. | 1.1.1.88 | ||||||||
| CAS no. | 37250-24-1 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
| hydroxymethylglutaryl-CoA reductase (NADPH) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 HMG-CoA reductase (NADPH), Human | |||||||||
| Identifiers | |||||||||
| EC no. | 1.1.1.34 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
HMG-CoA reductase is anchored in the membrane of the endoplasmic reticulum, and was long regarded as having seven transmembrane domains, with the active site located in a long carboxyl terminal domain in the cytosol. More recent evidence shows it to contain eight transmembrane domains.[5]
In humans, the gene for HMG-CoA reductase (NADPH) is located on the long arm of the fifth chromosome (5q13.3-14).[6] Related enzymes having the same function are also present in other animals, plants and bacteria.
Structure
editThe main isoform (isoform 1) of HMG-CoA reductase in humans is 888 amino acids long. It is a polytopic transmembrane protein (meaning it possesses many alpha helical transmembrane segments). It contains two main domains:
- a conserved N-terminal sterol-sensing domain (SSD, amino acid interval: 88–218). The related SSD of SCAP has been shown to bind cholesterol.[7][8]
- a C-terminal catalytic domain (amino acid interval: 489-871), namely the 3-hydroxy-3-methyl-glutaryl-CoA reductase domain. This domain is required for the proper enzymatic activity of the protein.[9]
Isoform 2 is 835 amino acids long. This variant is shorter because it lacks an exon in the middle region (amino acids 522–574). This does not affect any of the aforementioned domains.
Function
editHMGCR catalyses the conversion of HMG-CoA to mevalonic acid, a necessary step in the biosynthesis of cholesterol:
 |
Normally in mammalian cells this enzyme is competitively suppressed by cholesterol derived from the internalization and degradation of low density lipoprotein (LDL) via the LDL receptor as well as oxidized species of cholesterol. Competitive inhibitors of the reductase induce the expression of LDL receptors in the liver, which in turn increases the catabolism of plasma LDL and lowers the plasma concentration of cholesterol, which is considered, by those who accept the standard lipid hypothesis, an important determinant of atherosclerosis.[10] This enzyme is thus the target of the widely available cholesterol-lowering drugs known collectively as the statins (see Drugs section for more).
In Drosophila melanogaster, Hmgcr is the homolog of Human HMGCR, and plays crucial roles in regulating energy metabolism and food intake but also sleep homeostasis through the central mechanisms according to these studies
, https://www.mdpi.com/2073-4409/11/6/970 and https://www.mdpi.com/1424-8247/15/1/79
Interactive pathway map
editClick on genes, proteins and metabolites below to link to respective articles. [§ 1]
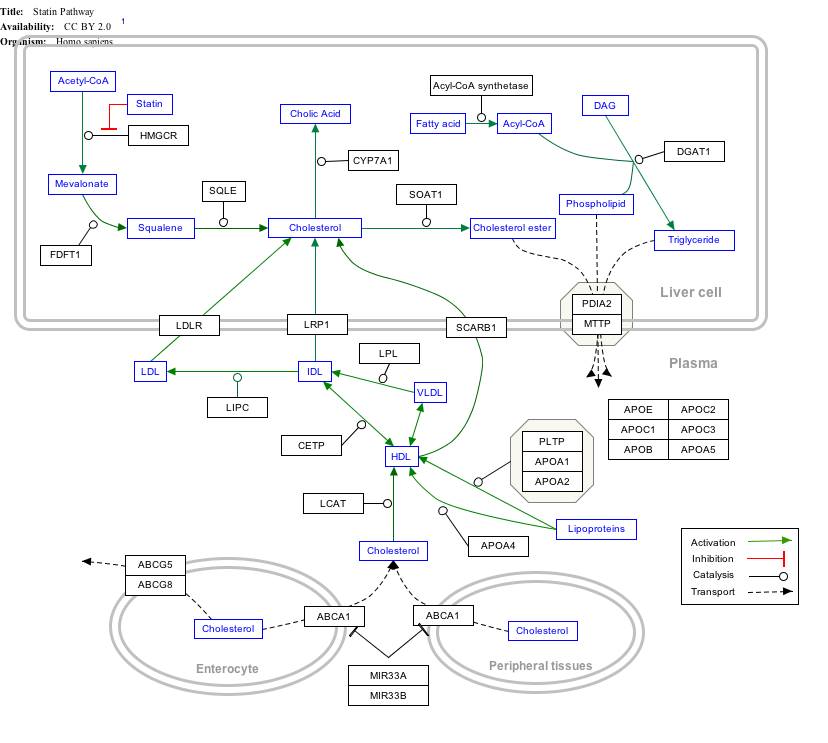
{kind=link}
- ^ The interactive pathway map can be edited at WikiPathways: "Statin_Pathway_WP430".
Inhibitors
editDrugs
editDrugs that inhibit HMG-CoA reductase, known collectively as HMG-CoA reductase inhibitors (or "statins"), are used to lower serum cholesterol as a means of reducing the risk for cardiovascular disease.[11]
These drugs include rosuvastatin (CRESTOR), lovastatin (Mevacor), atorvastatin (Lipitor), pravastatin (Pravachol), fluvastatin (Lescol), pitavastatin (Livalo), and simvastatin (Zocor).[12] Red yeast rice extract, one of the fungal sources from which the statins were discovered, contains several naturally occurring cholesterol-lowering molecules known as monacolins. The most active of these is monacolin K, or lovastatin (previously sold under the trade name Mevacor, and now available as generic lovastatin).[13]
Vytorin is drug that combines the use simvastatin and ezetimibe, which slows the formation of cholesterol by every cell in the body, along with ezetimibe reducing absorption of cholesterol, typically by about 53%, from the intestines.[14]
Statins, HMG-CoA reductase inhibitors, are competent in lowering cholesterol levels and reducing cardiac-related diseases. However, there have been controversies surrounding the potential of statins increasing the risk of new-onset diabetes mellitus (NOD). Experiments have demonstrated that glucose and cholesterol homeostasis are regulated by statins. The HMG-CoA reductase (HMGCR), converts HMG-CoA into mevalonic acid. Thus, when HMGCR activities are reduced, the cell associated cholesterols are also reduced. This results in the activation of SREBP-2-mediated signaling pathways. SREBP-2 activation for cholesterol homeostasis is crucial for the upregulation of low density lipoprotein (LDL) receptor (LDLR). The removal of LDL particles from blood circulation is enhanced when the number of LDLR on hepatocytes increases. Due to the removal of atherogenic lipoprotein particles, such as LDLs and intermediate density lipoproteins, HMGCR inhibitors have been proven to be efficient in reducing cardiovascular diseases from the blood circulation, which is represented by the reduction of LDL-cholesterol levels. In many studies, lipophilic statins are shown as more diabetogenic, possibly due to the fact that they can easily diffuse into cells and inhibit the production of isoprenoids which become more potent. Additionally, statins have been shown to change glucose levels as well.[15]
Hormones
editHMG-CoA reductase is active when blood glucose is high. The basic functions of insulin and glucagon are to maintain glucose homeostasis. Thus, in controlling blood sugar levels, they indirectly affect the activity of HMG-CoA reductase, but a decrease in activity of the enzyme is caused by AMP-activated protein kinase,[16] which responds to an increase in AMP concentration, and also to leptin.
Clinical significance
editSince the reaction catalysed by HMG-CoA reductase is the rate-limiting step in cholesterol synthesis, this enzyme represents the sole major drug target for contemporary cholesterol-lowering drugs in humans. The medical significance of HMG-CoA reductase has continued to expand beyond its direct role in cholesterol synthesis following the discovery that statins can offer cardiovascular health benefits independent of cholesterol reduction.[17] Statins have been shown to have anti-inflammatory properties,[18] most likely as a result of their ability to limit production of key downstream isoprenoids that are required for portions of the inflammatory response. It can be noted that blocking of isoprenoid synthesis by statins has shown promise in treating a mouse model of multiple sclerosis, an inflammatory autoimmune disease.[19]
Inhibition of HMG-CoA reductase by statins is lessened in patients with type 2 diabetes, which results in lessened inhibition of coronary atheromatous plaque, development.[20]
HMG-CoA reductase is an important developmental enzyme. Inhibition of its activity and the concomitant lack of isoprenoids that yields can lead to germ cell migration defects[21] as well as intracerebral hemorrhage.[22]
Homozygous mutation of HMGCR can lead to a form of limb girdle myopathy that may share features with mild statin-induced myopathy. The clinical syndrome was partially reversed in a model system by supplementation with the downstream metabolite mevalonolactone.[23]
The presence of anti HMG-CoA reductase antibodies is seen in people with statin-associated autoimmune myopathy, which is a very rare form of muscle damage caused by the immune system in people who take statin medications.[24] The exact mechanism is unclear. A combination of consistent findings on physical examination, the presence of anti HMG-CoA reductase antibodies in a person with myopathy, evidence of muscle breakdown, and muscle biopsy diagnose SAAM.[25]
Regulation
edit
Regulation of HMG-CoA reductase is achieved at several levels: transcription, translation, degradation and phosphorylation.
Transcription
editTranscription of the reductase gene is enhanced by the sterol regulatory element binding protein (SREBP). This protein binds to the sterol regulatory element (SRE), located on the 5' end of the reductase gene after controlled proteolytic processing. When SREBP is inactive, it is bound to the ER or nuclear membrane with another protein called SREBP cleavage-activating protein (SCAP). SCAP senses low cholesterol concentration and transports SREBP to the Golgi membrane where a consecutive proteolysis by S1P and S2P cleaves SREBP into an active nuclear form, nSREBP. nSREBPs migrate to the nucleus and activate transcription of SRE-containing genes. The nSREBP transcription factor is short-lived. When cholesterol levels rise, Insigs retains the SCAP-SREBP complex in the ER membrane by preventing its incorporation into COPII vesicles.[26][27]
Translation
editTranslation of mRNA is inhibited by a mevalonate derivative, which has been reported to be the isoprenoid farnesol,[28][29] although this role has been disputed.[30]
Degradation
editRising levels of sterols increase the susceptibility of the reductase enzyme to ER-associated degradation (ERAD) and proteolysis. Helices 2-6 (total of 8) of the HMG-CoA reductase transmembrane domain are thought to sense increased cholesterol levels (direct sterol binding to the SSD of HMG-CoA reductase has not been demonstrated). Lysine residues 89 and 248 can become ubiquinated by ER-resident E3 ligases. The identity of the multiple E3 ligases involved in HMG-CoA degradation is controversial, with suggested candidates being AMFR,[31] Trc8,[32] and RNF145[33][34] The involvement of AMFR and Trc8 has been contested.[35]
Phosphorylation
editShort-term regulation of HMG-CoA reductase is achieved by inhibition by phosphorylation (of Serine 872, in humans[36]). Decades ago it was believed that a cascade of enzymes controls the activity of HMG-CoA reductase: an HMG-CoA reductase kinase was thought to inactivate the enzyme, and the kinase in turn was held to be activated via phosphorylation by HMG-CoA reductase kinase kinase. An excellent review on regulation of the mevalonate pathway by Nobel Laureates Joseph Goldstein and Michael Brown adds specifics: HMG-CoA reductase is phosphorylated and inactivated by an AMP-activated protein kinase, which also phosphorylates and inactivates acetyl-CoA carboxylase, the rate-limiting enzyme of fatty acid biosynthesis.[37] Thus, both pathways utilizing acetyl-CoA for lipid synthesis are inactivated when energy charge is low in the cell, and concentrations of AMP rise. There has been a great deal of research on the identity of upstream kinases that phosphorylate and activate the AMP-activated protein kinase.[38]
Fairly recently, LKB1 has been identified as a likely AMP kinase kinase,[39] which appears to involve calcium/calmodulin signaling. This pathway likely transduces signals from leptin, adiponectin, and other signaling molecules.[38]
See also
editReferences
edit- ^ a b c GRCh38: Ensembl release 89: ENSG00000113161 – Ensembl, May 2017
- ^ a b c GRCm38: Ensembl release 89: ENSMUSG00000021670 – Ensembl, May 2017
- ^ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ Roitelman J, Olender EH, Bar-Nun S, Dunn WA, Simoni RD (June 1992). "Immunological evidence for eight spans in the membrane domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase: implications for enzyme degradation in the endoplasmic reticulum". The Journal of Cell Biology. 117 (5): 959–973. doi:10.1083/jcb.117.5.959. PMC 2289486. PMID 1374417.
- ^ Lindgren V, Luskey KL, Russell DW, Francke U (December 1985). "Human genes involved in cholesterol metabolism: chromosomal mapping of the loci for the low density lipoprotein receptor and 3-hydroxy-3-methylglutaryl-coenzyme A reductase with cDNA probes". Proceedings of the National Academy of Sciences of the United States of America. 82 (24): 8567–8571. Bibcode:1985PNAS...82.8567L. doi:10.1073/pnas.82.24.8567. PMC 390958. PMID 3866240.
- ^ Brown MS, Radhakrishnan A, Goldstein JL (June 2018). "Retrospective on Cholesterol Homeostasis: The Central Role of Scap". Annual Review of Biochemistry. 87: 783–807. doi:10.1146/annurev-biochem-062917-011852. PMC 5828883. PMID 28841344.
- ^ Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL (July 2004). "Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain". Molecular Cell. 15 (2): 259–268. doi:10.1016/j.molcel.2004.06.019. PMID 15260976.
- ^ Costa CH, Oliveira AR, Dos Santos AM, da Costa KS, Lima AH, Alves CN, Lameira J (October 2019). "Computational study of conformational changes in human 3-hydroxy-3-methylglutaryl coenzyme reductase induced by substrate binding". Journal of Biomolecular Structure & Dynamics. 37 (16): 4374–4383. doi:10.1080/07391102.2018.1549508. PMID 30470158. S2CID 53717806.
- ^ "Entrez Gene: HMGCR 3-hydroxy-3-methylglutaryl-Coenzyme A reductase".
- ^ Farmer JA (1998). "Aggressive lipid therapy in the statin era". Progress in Cardiovascular Diseases. 41 (2): 71–94. doi:10.1016/S0033-0620(98)80006-6. PMID 9790411.
- ^ "Is there a "best" statin drug?". The Johns Hopkins Medical Letter Health After 50. 15 (11): 4–5. January 2004. PMID 14983817.
- ^ Lin YL, Wang TH, Lee MH, Su NW (January 2008). "Biologically active components and nutraceuticals in the Monascus-fermented rice: a review" (PDF). Applied Microbiology and Biotechnology. 77 (5): 965–973. doi:10.1007/s00253-007-1256-6. PMID 18038131. S2CID 33299544.
- ^ Flores NA (September 2004). "Ezetimibe + simvastatin (Merck/Schering-Plough)". Current Opinion in Investigational Drugs. 5 (9): 984–992. PMID 15503655.
- ^ Han KH (November 2018). "Functional Implications of HMG-CoA Reductase Inhibition on Glucose Metabolism". Korean Circulation Journal. 48 (11). The Korean Society of Cardiology: 951–963. doi:10.4070/kcj.2018.0307. PMC 6196158. PMID 30334382.
- ^ Hardie DG (February 1992). "Regulation of fatty acid and cholesterol metabolism by the AMP-activated protein kinase". Biochimica et Biophysica Acta (BBA) - Lipids and Lipid Metabolism. 1123 (3): 231–238. doi:10.1016/0005-2760(92)90001-c. PMID 1536860.
- ^ Arnaud C, Veillard NR, Mach F (April 2005). "Cholesterol-independent effects of statins in inflammation, immunomodulation and atherosclerosis". Current Drug Targets. Cardiovascular & Hematological Disorders. 5 (2): 127–134. doi:10.2174/1568006043586198. PMID 15853754.
- ^ Sorrentino S, Landmesser U (December 2005). "Nonlipid-lowering effects of statins". Current Treatment Options in Cardiovascular Medicine. 7 (6): 459–466. doi:10.1007/s11936-005-0031-1. PMID 16283973. S2CID 44918429.
- ^ Stüve O, Youssef S, Steinman L, Zamvil SS (June 2003). "Statins as potential therapeutic agents in neuroinflammatory disorders". Current Opinion in Neurology. 16 (3): 393–401. doi:10.1097/00019052-200306000-00021. PMID 12858078.
- ^ Mashayekhi-Sardoo H, Atkin SL, Montecucco F, Sahebkar A (2021). "Potential Alteration of Statin-Related Pharmacological Features in Diabetes Mellitus". BioMed Research International. 2021: 6698743. doi:10.1155/2021/6698743. PMC 8018846. PMID 33834073.
- ^ Thorpe JL, Doitsidou M, Ho SY, Raz E, Farber SA (February 2004). "Germ cell migration in zebrafish is dependent on HMGCoA reductase activity and prenylation". Developmental Cell. 6 (2): 295–302. doi:10.1016/S1534-5807(04)00032-2. hdl:11858/00-001M-0000-0012-EE5B-7. PMID 14960282.
- ^ Eisa-Beygi S, Hatch G, Noble S, Ekker M, Moon TW (January 2013). "The 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) pathway regulates developmental cerebral-vascular stability via prenylation-dependent signalling pathway". Developmental Biology. 373 (2): 258–266. doi:10.1016/j.ydbio.2012.11.024. PMID 23206891.
- ^ Yogev Y, Shorer Z, Koifman A, Wormser O, Drabkin M, Halperin D, Dolgin V, Proskorovski-Ohayon R, Hadar N, Davidov G, Nudelman H, Zarivach R, Shelef I, Perez Y, Birk OS (February 2023). "Limb girdle muscular disease caused by HMGCR mutation and statin myopathy treatable with mevalonolactone". Proc Natl Acad Sci U S A. 120 (7): e2217831120. Bibcode:2023PNAS..12017831Y. doi:10.1073/pnas.2217831120. PMC 9963716. PMID 36745799.
- ^ Thompson PD, Panza G, Zaleski A, Taylor B (May 2016). "Statin-Associated Side Effects". Journal of the American College of Cardiology (Review). 67 (20): 2395–2410. doi:10.1016/j.jacc.2016.02.071. PMID 27199064.
- ^ Mammen AL (February 2016). "Statin-Associated Autoimmune Myopathy". The New England Journal of Medicine (Review). 374 (7): 664–669. doi:10.1056/NEJMra1515161. PMID 26886523.
- ^ Sun LP, Seemann J, Goldstein JL, Brown MS (April 2007). "Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins". Proceedings of the National Academy of Sciences of the United States of America. 104 (16): 6519–6526. Bibcode:2007PNAS..104.6519S. doi:10.1073/pnas.0700907104. PMC 1851663. PMID 17428919.
- ^ Sun LP, Li L, Goldstein JL, Brown MS (July 2005). "Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro". The Journal of Biological Chemistry. 280 (28): 26483–26490. doi:10.1074/jbc.M504041200. PMID 15899885.
- ^ Meigs TE, Roseman DS, Simoni RD (April 1996). "Regulation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase degradation by the nonsterol mevalonate metabolite farnesol in vivo". The Journal of Biological Chemistry. 271 (14): 7916–7922. doi:10.1074/jbc.271.14.7916. PMID 8626470.
- ^ Meigs TE, Simoni RD (September 1997). "Farnesol as a regulator of HMG-CoA reductase degradation: characterization and role of farnesyl pyrophosphatase". Archives of Biochemistry and Biophysics. 345 (1): 1–9. doi:10.1006/abbi.1997.0200. PMID 9281305.
- ^ Keller RK, Zhao Z, Chambers C, Ness GC (April 1996). "Farnesol is not the nonsterol regulator mediating degradation of HMG-CoA reductase in rat liver". Archives of Biochemistry and Biophysics. 328 (2): 324–330. doi:10.1006/abbi.1996.0180. PMID 8645011.
- ^ Song BL, Sever N, DeBose-Boyd RA (September 2005). "Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase". Molecular Cell. 19 (6): 829–840. doi:10.1016/j.molcel.2005.08.009. PMID 16168377.
- ^ Jo Y, Lee PC, Sguigna PV, DeBose-Boyd RA (December 2011). "Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8". Proceedings of the National Academy of Sciences of the United States of America. 108 (51): 20503–20508. Bibcode:2011PNAS..10820503J. doi:10.1073/pnas.1112831108. PMC 3251157. PMID 22143767.
- ^ Jiang LY, Jiang W, Tian N, Xiong YN, Liu J, Wei J, et al. (March 2018). "Ring finger protein 145 (RNF145) is a ubiquitin ligase for sterol-induced degradation of HMG-CoA reductase". The Journal of Biological Chemistry. 293 (11): 4047–4055. doi:10.1074/jbc.RA117.001260. PMC 5857978. PMID 29374057.
- ^ Menzies SA, Volkmar N, van den Boomen DJ, Timms RT, Dickson AS, Nathan JA, Lehner PJ (December 2018). "The sterol-responsive RNF145 E3 ubiquitin ligase mediates the degradation of HMG-CoA reductase together with gp78 and Hrd1". eLife. 7. doi:10.7554/eLife.40009. PMC 6292692. PMID 30543180.
- ^ Tsai YC, Leichner GS, Pearce MM, Wilson GL, Wojcikiewicz RJ, Roitelman J, Weissman AM (December 2012). "Differential regulation of HMG-CoA reductase and Insig-1 by enzymes of the ubiquitin-proteasome system". Molecular Biology of the Cell. 23 (23): 4484–4494. doi:10.1091/mbc.E12-08-0631. PMC 3510011. PMID 23087214.
- ^ Istvan ES, Palnitkar M, Buchanan SK, Deisenhofer J (March 2000). "Crystal structure of the catalytic portion of human HMG-CoA reductase: insights into regulation of activity and catalysis". The EMBO Journal. 19 (5): 819–830. doi:10.1093/emboj/19.5.819. PMC 305622. PMID 10698924.
- ^ Goldstein JL, Brown MS (February 1990). "Regulation of the mevalonate pathway". Nature. 343 (6257): 425–430. Bibcode:1990Natur.343..425G. doi:10.1038/343425a0. PMID 1967820. S2CID 30477478.
- ^ a b Hardie DG, Scott JW, Pan DA, Hudson ER (July 2003). "Management of cellular energy by the AMP-activated protein kinase system". FEBS Letters. 546 (1): 113–120. Bibcode:2003FEBSL.546..113H. doi:10.1016/S0014-5793(03)00560-X. PMID 12829246. S2CID 42881381.
- ^ Witters LA, Kemp BE, Means AR (January 2006). "Chutes and Ladders: the search for protein kinases that act on AMPK". Trends in Biochemical Sciences. 31 (1): 13–16. doi:10.1016/j.tibs.2005.11.009. PMID 16356723.
Further reading
edit- Hodge VJ, Gould SJ, Subramani S, Moser HW, Krisans SK (December 1991). "Normal cholesterol synthesis in human cells requires functional peroxisomes". Biochemical and Biophysical Research Communications. 181 (2): 537–541. doi:10.1016/0006-291X(91)91222-X. PMID 1755834.
- Ramharack R, Tam SP, Deeley RG (November 1990). "Characterization of three distinct size classes of human 3-hydroxy-3-methylglutaryl coenzyme A reductase mRNA: expression of the transcripts in hepatic and nonhepatic cells". DNA and Cell Biology. 9 (9): 677–690. doi:10.1089/dna.1990.9.677. PMID 1979742.
- Clarke PR, Hardie DG (August 1990). "Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver". The EMBO Journal. 9 (8): 2439–2446. doi:10.1002/j.1460-2075.1990.tb07420.x. PMC 552270. PMID 2369897.
- Luskey KL, Stevens B (August 1985). "Human 3-hydroxy-3-methylglutaryl coenzyme A reductase. Conserved domains responsible for catalytic activity and sterol-regulated degradation". The Journal of Biological Chemistry. 260 (18): 10271–10277. doi:10.1016/S0021-9258(17)39242-6. PMID 2991281.
- Humphries SE, Tata F, Henry I, Barichard F, Holm M, Junien C, Williamson R (1986). "The isolation, characterisation, and chromosomal assignment of the gene for human 3-hydroxy-3-methylglutaryl coenzyme A reductase, (HMG-CoA reductase)". Human Genetics. 71 (3): 254–258. doi:10.1007/BF00284585. PMID 2998972. S2CID 10619592.
- Beg ZH, Stonik JA, Brewer HB (September 1987). "Phosphorylation and modulation of the enzymic activity of native and protease-cleaved purified hepatic 3-hydroxy-3-methylglutaryl-coenzyme A reductase by a calcium/calmodulin-dependent protein kinase". The Journal of Biological Chemistry. 262 (27): 13228–13240. doi:10.1016/S0021-9258(18)45191-5. PMID 3308873.
- Osborne TF, Goldstein JL, Brown MS (August 1985). "5' end of HMG CoA reductase gene contains sequences responsible for cholesterol-mediated inhibition of transcription". Cell. 42 (1): 203–212. doi:10.1016/S0092-8674(85)80116-1. PMID 3860301. S2CID 37319421.
- Lindgren V, Luskey KL, Russell DW, Francke U (December 1985). "Human genes involved in cholesterol metabolism: chromosomal mapping of the loci for the low density lipoprotein receptor and 3-hydroxy-3-methylglutaryl-coenzyme A reductase with cDNA probes". Proceedings of the National Academy of Sciences of the United States of America. 82 (24): 8567–8571. Bibcode:1985PNAS...82.8567L. doi:10.1073/pnas.82.24.8567. PMC 390958. PMID 3866240.
- Lehoux JG, Kandalaft N, Belisle S, Bellabarba D (October 1985). "Characterization of 3-hydroxy-3-methylglutaryl coenzyme A reductase in human adrenal cortex". Endocrinology. 117 (4): 1462–1468. doi:10.1210/endo-117-4-1462. PMID 3896758.
- Boguslawski W, Sokolowski W (1984). "HMG-CoA reductase activity in the microsomal fraction from human placenta in early and term pregnancy". The International Journal of Biochemistry. 16 (9): 1023–1026. doi:10.1016/0020-711X(84)90120-4. PMID 6479432.
- Harwood HJ, Schneider M, Stacpoole PW (September 1984). "Measurement of human leukocyte microsomal HMG-CoA reductase activity". Journal of Lipid Research. 25 (9): 967–978. doi:10.1016/S0022-2275(20)37733-6. PMID 6491541.
- Nguyen LB, Salen G, Shefer S, Bullock J, Chen T, Tint GS, et al. (July 1994). "Deficient ileal 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in sitosterolemia: sitosterol is not a feedback inhibitor of intestinal cholesterol biosynthesis". Metabolism. 43 (7): 855–859. doi:10.1016/0026-0495(94)90266-6. PMID 8028508.
- Bennis F, Favre G, Le Gaillard F, Soula G (October 1993). "Importance of mevalonate-derived products in the control of HMG-CoA reductase activity and growth of human lung adenocarcinoma cell line A549". International Journal of Cancer. 55 (4): 640–645. doi:10.1002/ijc.2910550421. PMID 8406993. S2CID 23842867.
- Van Doren M, Broihier HT, Moore LA, Lehmann R (December 1998). "HMG-CoA reductase guides migrating primordial germ cells". Nature. 396 (6710): 466–469. Bibcode:1998Natur.396..466V. doi:10.1038/24871. PMID 9853754. S2CID 4430351.
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, et al. (July 1999). "Characterization of single-nucleotide polymorphisms in coding regions of human genes". Nature Genetics. 22 (3): 231–238. doi:10.1038/10290. PMID 10391209. S2CID 195213008.
- Aboushadi N, Engfelt WH, Paton VG, Krisans SK (September 1999). "Role of peroxisomes in isoprenoid biosynthesis". The Journal of Histochemistry and Cytochemistry. 47 (9): 1127–1132. doi:10.1177/002215549904700904. PMID 10449533. S2CID 1596555.
- Honda A, Salen G, Honda M, Batta AK, Tint GS, Xu G, et al. (February 2000). "3-Hydroxy-3-methylglutaryl-coenzyme A reductase activity is inhibited by cholesterol and up-regulated by sitosterol in sitosterolemic fibroblasts". The Journal of Laboratory and Clinical Medicine. 135 (2): 174–179. doi:10.1067/mlc.2000.104459. PMID 10695663.
- Istvan ES, Deisenhofer J (May 2001). "Structural mechanism for statin inhibition of HMG-CoA reductase". Science. 292 (5519): 1160–1164. Bibcode:2001Sci...292.1160I. doi:10.1126/science.1059344. PMID 11349148. S2CID 37686043.
- Rasmussen LM, Hansen PR, Nabipour MT, Olesen P, Kristiansen MT, Ledet T (December 2001). "Diverse effects of inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase on the expression of VCAM-1 and E-selectin in endothelial cells". The Biochemical Journal. 360 (Pt 2): 363–370. doi:10.1042/0264-6021:3600363. PMC 1222236. PMID 11716764.
External links
edit- Cholesterol Synthesis Archived 4 July 2017 at the Wayback Machine - has some good regulatory details
- Proteopedia HMG-CoA_Reductase - the HMG-CoA Reductase Structure in Interactive 3D
- Overview of all the structural information available in the PDB for UniProt: P04035 (3-hydroxy-3-methylglutaryl-coenzyme A reductase) at the PDBe-KB.